


Super-Enhancer-Driven SOX4/SMAD3 Mediate Membrane Remodeling by Regulating Phospholipid Metabolism to Accelerate Leukemia Progression
《Advanced Science》(IF=14.1)
摘要
慢性髓系白血病(CML)由 BCR -ABL融合癌基因驱动,病程从慢性期(CP)进展至急变期(BP)。
虽然酪氨酸激酶抑制剂(TKIs)能有效控制CML-CP,但CML-BP仍是一个治疗难题,其特征是治疗耐药性和生存率低下。
越来越多的证据表明,表观遗传调控元件的异常激活会重塑癌症中的转录组,导致对特定转录调节因子产生依赖性,从而推动癌症进展。
然而,具体哪些转录机制促进了CML-CP向CML-BP的转变尚不明确。本研究在CML-BP中鉴定出由超级增强子驱动的转录因子SOX4和SMAD3。
SOX4与SMAD3通过分别结合各自的超级增强子和启动子,形成正反馈轴。功能实验证实该轴在体外和体内均能促进白血病进展。
从机制上看,SOX4/SMAD3结合于受体酪氨酸激酶 AXL 的启动子和增强子区域,增强其转录活性,进而激活 AKT /ERK/STAT5信号通路;同时,它们通过转录上调LPCAT1基因表达,后者可重塑膜磷脂结构以促进 AXL 的定位。
值得注意的是, AXL 抑制剂贝森替尼在体内外模型中均能有效抑制CML-BP的进展。
总体而言,我们的研究结果确立了SE驱动的SOX4和SMAD3作为CML-BP的关键调控因子,并将贝森替尼确定为一种前景广阔的治疗策略。
一、研究背景与科学问题
1.CML疾病特点
由t(9;22)易位产生BCR-ABL融合癌基因驱动,分三期:慢性期(CP,原始细胞<10%)、加速期(AP)、急变期(BP,原始细胞≥20%)。
急变后呈急性白血病表型,TKIs(伊马替尼等)普遍耐药,生存期极短,是临床未满足需求。
2.表观遗传与超级增强子(SE)
SE是高密度增强子簇,高H3K27ac修饰,驱动癌基因高表达,决定肿瘤身份与进展。SE在肉瘤、淋巴瘤、神经母细胞瘤中已被证实,但在CML急变中的驱动机制完全未知。
3. 核心科学问题
CML急变是否存在SE驱动的关键转录因子?
其如何通过非BCR-ABL依赖通路推动急变?
能否找到可靶向的关键节点用于治疗?
二、研究策略与技术路线
1.临床队列:
40例CML(19例CP,21例BP)原代样本。
2.组学技术
◦H3K27ac ChIP-seq(鉴定SE)
◦RNA-seq(表达谱)
◦CUT&Tag(SOX4/SMAD3全基因组结合)
◦脂质组学(磷脂代谢)
3C(染色质环)、ChIP-re-ChIP、双荧光素酶、Co-IP等。
3.细胞模型:
K562、LAMA-84、KBM5、KU812、KBM5-T315I(耐药株)。
4.动物模型
◦BA模型:BCR-ABL单驱动
◦BA/NH模型:BCR-ABL+NUP98-HOXA9共驱动(更接近人急变)
5.干预手段:
shRNA敲低、过表达、dCas9-KRAB沉默增强子、药物抑制剂。
三、结果
3.1 鉴定CML急变期SE驱动的SOX4/SMAD3正反馈轴
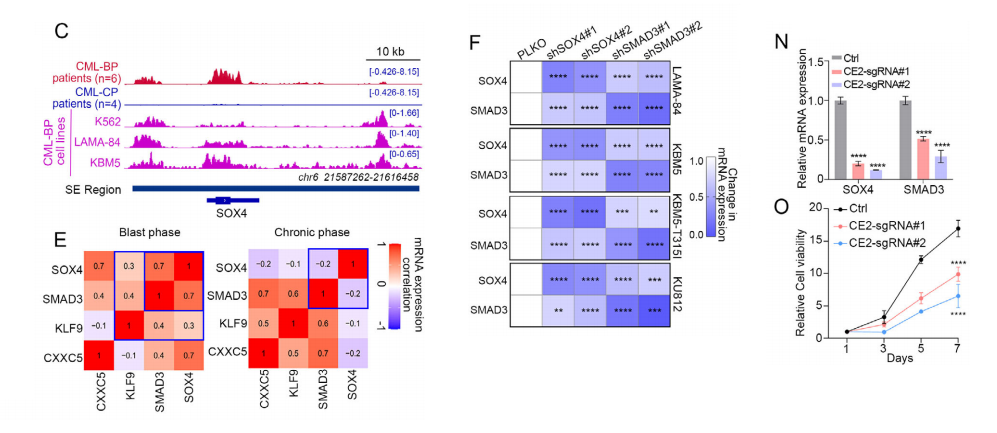
1.筛选SE驱动转录因子
•对比CP与BP的H3K27ac ChIP-seq,鉴定24个BP特异性SE。
• 结合RNA-seq,筛选出4个显著上调的SE驱动转录因子:SOX4、SMAD3、CXXC5、KLF9。
• IGV显示:BP样本在SOX4、SMAD3基因座的SE区域H3K27ac信号显著高于CP。
2. SOX4与SMAD3呈急变期特异性正相关• TCGA与本队列表达数据:仅在CML-BP强正相关,CP与AML中无此相关性。
• 细胞水平验证:◦ 敲低SOX4 → SMAD3表达下降;敲低SMAD3 → SOX4表达下降。◦ 过表达SOX4 → SMAD3上升;过表达SMAD3 → SOX4上升。
• BET抑制剂(ABBV-744、ARV-771)可剂量依赖性抑制两者,证明依赖SE活性。
3. 相互调控的分子基础
• 基序分析:SOX4与SMAD3的SE区均含对方结合基序。
• CUT&Tag + ChIP-re-ChIP:两者共结合在彼此的启动子与SE区。
• 蛋白互作(Co-IP):SOX4与SMAD3直接结合。
• 3C实验:SOX4的增强子CE2与启动子形成染色质环,空间上促进转录。
• dCas9-KRAB沉默CE2:H3K27ac下降、两者结合减少、表达下降、细胞增殖受抑、分化恢复。
结论:SOX4/SMAD3形成SE驱动的自我激活+相互激活正反馈环路,是CML急变的核心转录调控模块。
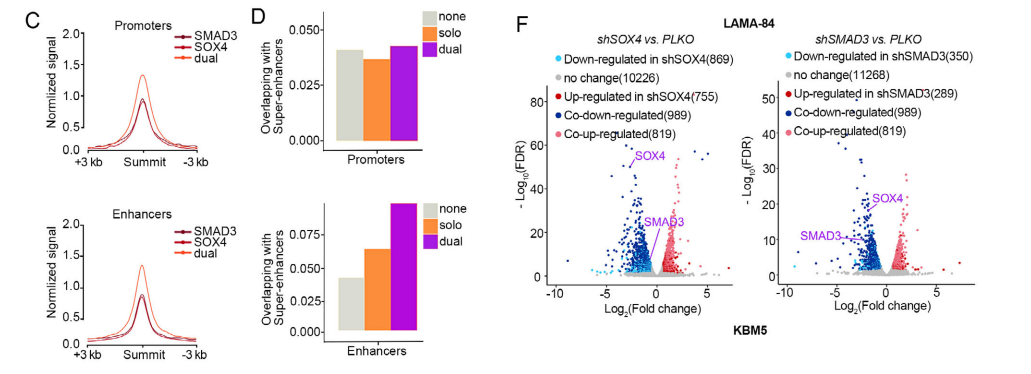
3.2 SOX4/SMAD3协同重塑转录组驱动急变1. 全基因组协同结合特征
• 两者更倾向共结合(dual-binding),而非单独结合。
• 共结合区域:H3K27ac更高、更富集SE、靶基因表达更高。
• 约40%共结合区域中,两者结合位点间距<146 bp(核小体跨度),保证高效协同。
2. 敲低后转录组变化
• 敲低SOX4或SMAD3,差异基因高度重叠。• GSEA显示:共同下调基因显著富集于CML急变期特征基因集。
• 共同上调基因富集于分化相关通路。
结论:SOX4/SMAD3以高度协同方式构建急变特异性转录程序。
3.3 SOX4/SMAD3功能上驱动CML恶性进展(体外)
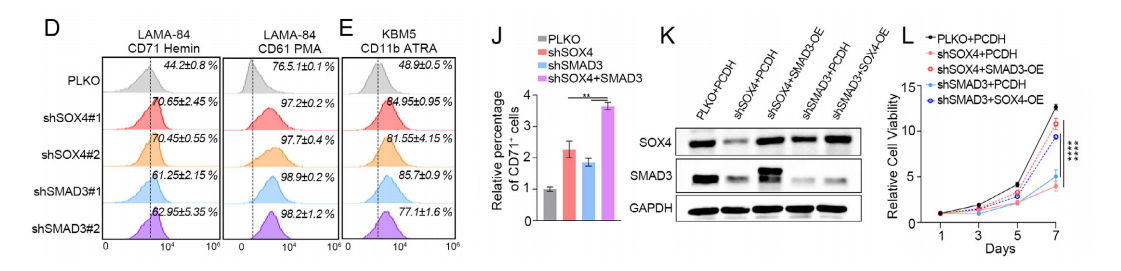
1.增殖与集落形成
• 单敲低SOX4/SMAD3:细胞增殖减慢、集落数量减少40%–60%、体积变小。
• 过表达:增殖显著增强。
• 双敲低:抑制效果显著强于单敲低,呈协同效应。
2. 细胞分化
• 敲低:促进红系(CD71)、巨核系(CD61)、粒系(CD11b)分化。
• 过表达:抑制分化,维持“未成熟急变表型”。
3. 回补实验
• 敲低SOX4 → 过表达SMAD3可部分恢复恶性表型。
• 敲低SMAD3 → 过表达SOX4可部分恢复恶性表型。
结论:SOX4/SMAD3是CML急变的必需因子,协同促进增殖、抑制分化、驱动急变。
3.4 下游通路——AXL是非BCR-ABL依赖的关键效应激酶
1.通路富集
• 共同下调基因富集:受体酪氨酸激酶(RTK)信号、脂质代谢。
• 排除BCR-ABL:敲低SOX4/SMAD3不改变BCR-ABL表达与磷酸化。
2. 锁定AXL
• 多个RTK中,仅AXL被两者共同调控。• SOX4/SMAD3直接结合AXL启动子与增强子,转录激活AXL。
3. AXL功能验证
• 敲低AXL:抑制增殖、促进分化,与SOX4/SMAD3敲低表型一致。
• AXL抑制剂Bemcentinib:有效杀伤CML-BP细胞,包括T315I耐药株。
• 信号机制:AXL激活p-AKT、p-ERK、p-STAT5,构成致癌信号轴。
4. 配体GAS6作用
• 内源性GAS6不受SOX4/SMAD3/AXL影响。• 外源性GAS6仅微弱回补表型,提示AXL膜定位比配体更关键。
结论:SOX4/SMAD3通过AXL激活非BCR-ABL依赖的致癌信号。
3.5 膜代谢机制——LPCAT1重塑磷脂决定AXL膜定位与活性
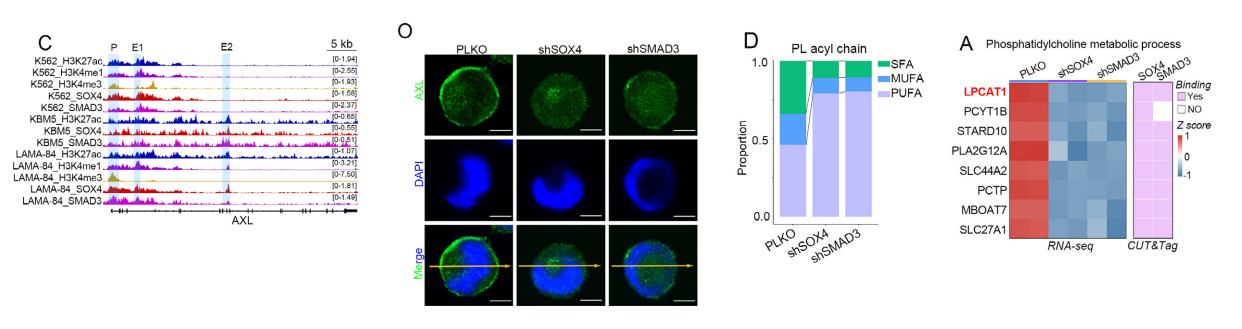
1.脂质组学发现
• 敲低SOX4/SMAD3:饱和磷脂(SFA-PL)显著下降,多不饱和磷脂(PUFA-PL)上升。
• 关键分子:DPPC(16:0/16:0) 下降最显著。
2. 膜流动性与AXL定位
• FRAP实验:敲低后膜流动性升高。
• 免疫荧光:AXL从细胞膜转位到胞内,失去活性。
3. 关键酶LPCAT1
• SOX4/SMAD3直接结合LPCAT1启动子与增强子,转录激活。
• LPCAT1是磷脂重塑(Lands循环)关键酶,负责合成饱和磷脂酰胆碱(PC)。
• 敲低LPCAT1:
◦ 饱和磷脂下降、膜流动性上升◦ AXL内吞、信号失活
◦ 增殖受抑、分化恢复
• 回补实验:
◦ 过表达LPCAT1 → 恢复膜流动性、AXL膜定位、增殖能力
◦ 外源添加DPPC → 同样可回补表型
4. 双机制协同
• SOX4/SMAD3对CML进展的驱动依赖两条通路缺一不可:
1.直接转录上调AXL
2. 通过LPCAT1-饱和磷脂重塑膜,保证AXL定位与活化
• 仅回补AXL / 仅回补LPCAT1 / 仅加DPPC,均不能完全逆转;三者联合才可完全恢复。
结论:SOX4/SMAD3-LPCAT1-饱和磷脂轴通过膜重塑控制AXL功能,是此前未被发现的“代谢-信号”耦联机制。
3.6 体内验证——靶向该轴可抑制CML急变并延长生存
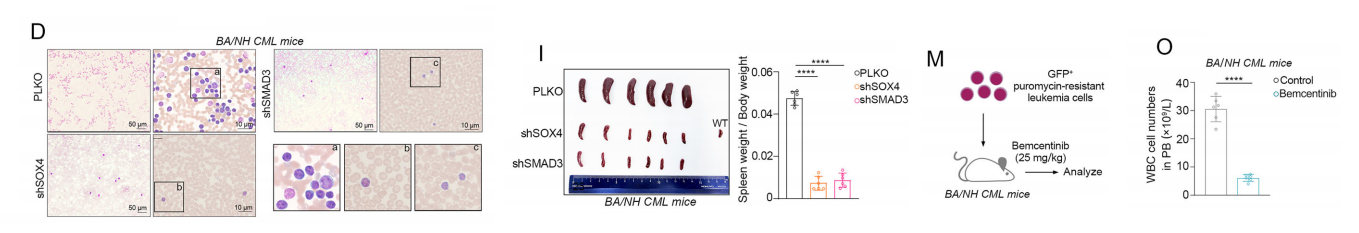
1.敲低SOX4/SMAD3的治疗效果
• 外周血:GFP+白血病细胞减少、白细胞总数下降。
• 骨髓:白血病细胞植入降低、原始细胞比例下降、分化恢复。
• 脾脏:脾肿大减轻、白血病浸润减少。
• 生存:显著延长小鼠生存期。
• 分子验证:体内同样下调AXL、LPCAT1,磷脂饱和度下降。
2. AXL抑制剂Bemcentinib治疗效果
• 给药方式:25 mg/kg,每日两次灌胃。
• 疗效:降低白血病负荷、改善血象、减轻脾肿大、延长生存。
• 安全性:体重无明显下降,肝肾功能(ALT/AST/CR)无异常。
• 信号:显著抑制p-AXL、p-STAT5、p-AKT、p-ERK。
3. 联合用药潜力
• Bemcentinib与伊马替尼具有强协同作用,提示可克服TKIs耐药。
四、结论
1.CML急变期出现超级增强子激活,驱动SOX4与SMAD3高表达。
2. SOX4与SMAD3形成正反馈环路,稳定维持高表达。
3. 双重机制驱动急变:
◦转录轴:直接激活AXL,启动AKT/ERK/STAT5致癌信号。
◦ 代谢轴:激活LPCAT1,提高饱和磷脂水平,降低膜流动性,保证AXL定位于细胞膜并充分活化。
4. AXL抑制剂Bemcentinib可有效阻断该轴,抑制CML急变,是潜在临床药物。
5. 该研究首次建立:SE→转录因子→磷脂代谢→膜受体定位→致癌信号的完整链条,为白血病急变机制提供全新范式。
五、创新点与临床价值
1.首次报道SE驱动的SOX4/SMAD3是CML急变的核心转录因子。
2. 首次揭示LPCAT1-饱和磷脂-膜流动性调控RTK定位的代谢-信号耦联机制。
3. 首次证实AXL抑制剂Bemcentinib对CML急变有效,可克服耐药。
4. 为CML急变提供3个可靶向节点:SOX4/SMAD3、LPCAT1、AXL。
六、总论
本研究揭示了一个由SOX4/SMAD3协同作用驱动的特异性转录调控网络:SOX4与SMAD3不仅直接调控 AXL 的表达,还通过调节LPCAT1介导的膜磷脂重塑影响其膜定位与激活——这两项机制在慢性髓系白血病(CML)进展中均起关键作用。这些发现深化了我们对CML转化过程中转录失调机制的理解,并为潜在治疗策略提供了依据。

作者使用伯信生物明星产品3C试剂盒进行分子调控机制研究。
伯信好物推荐
Chromosome Conformation Capture(3C) Kit
产品介绍
染色体构象捕获技术(chromosome conformation capture,3C),是革新性的绘制染色质相互作用的工具,应用于酵母中研究基因表达时,染色质的空间构象分析,继而发展为在后生动物中研究细胞内染色质间的相互作用。
3C 技术,能从全基因组的角度诠释了蛋白质因子与染色质相互作用的关系,以及细胞核内互作染色质的空间构象。

伯信 3C Kit分为:
Bes5006 Chromosome Conformation Capture (3C) kit 12T
Bes5006 Chromosome Conformation Capture (3C) kit 40T
实验原理:
利用甲醛固定细胞内的染色质,然后通过限制性内切酶打断DNA,采用连接酶使得被交联在一起的DNA片段之间能够发生邻位连接,然后通过在两个基因的酶切位点附近设计引物。
若两者有相互作用,则在酶切和连接处理之后,两者的线性位置会距离很近,可以通过PCR反应扩增出来,若两者无相互作用,则经过处理后线性位置仍然相距很远,不能被扩增出来。
技术流程:

结果实例:

产品优势:
1.操作简便,结果准确可靠
2.配备完整
3.伯信独立研发

